 thanks to @Department of Biology Memorial University of Newfoundland
thanks to @Department of Biology Memorial University of Newfoundland
{kind=link}
January 2019
Introduction
Welcome back RSG friends! Christmas is almost a dear memory and we are full of energy to start this new year. As a special topic this month we revise three papers related to quantitative approaches to study transcription. We hope you appreciate this topic, have fun and keep in touch. See you next month! Yours:
RGS Germany (ISCB Student Council) – Tommaso Andreani, Neetika Nath, Nikos Papadopoulos, Yvonne Gladbach
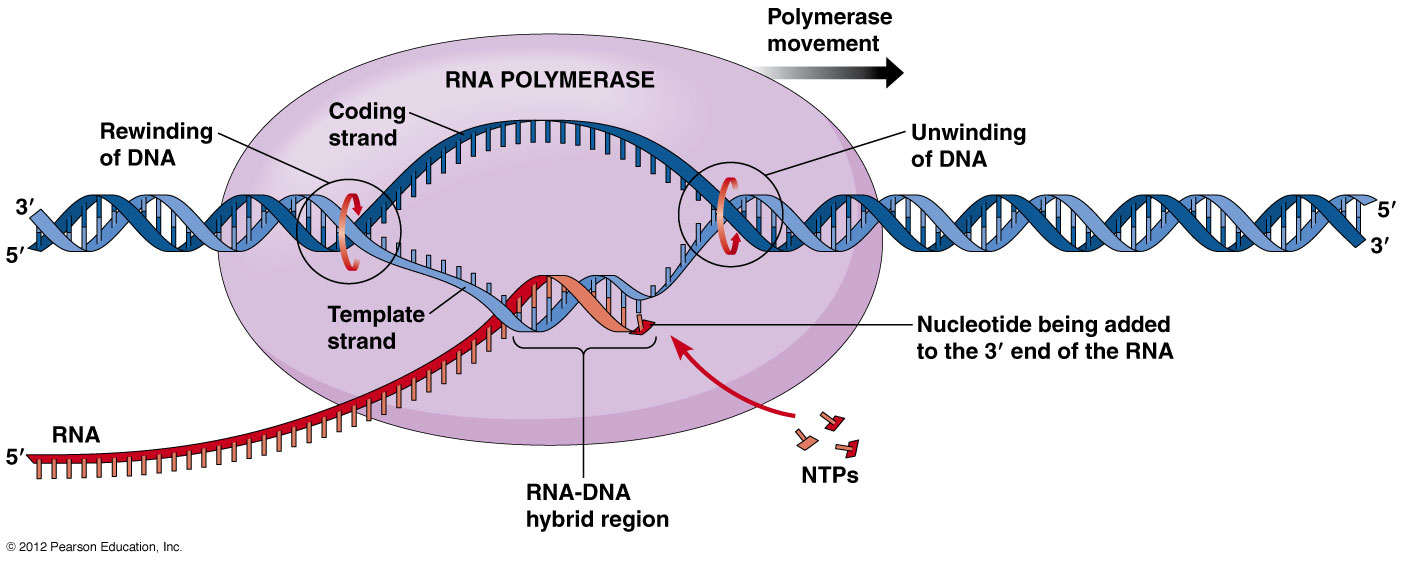
A unified approach for quantifying and interpreting DNA shape readout by transcription factors
The study of transcription with quantitative approaches is a key element in the transcriptomic analysis. Transcription factors (TFs) interpret DNA sequence based on each nucleotide polymer by probing its chemical and structural properties. In addition stands the DNA shape, which is a limited representation of dependencies of the nucleotides. Any mechanism-agnostic protein-DNA binding model can be analyzed using a unified mathematical representation of the DNA sequence dependence of shape and TF binding.
The sensitivity of TF binding to subtle deviations from one of the possible double-helix structures, B-DNA, is the mechanism called DNA shape readout. In the case TF binding specificity data are available, the DNA shape readout can only be acquired by dissecting the specificity in terms of base and shape readout.
Parameterizing base readout using a scoring matrix is an established method and it takes the corresponding contribution to the binding affinity into account. The computation of it is based on the identity indicators along the binding site. On the other hand, the shape readout is another established method that takes a profile of shape-sensitivity coefficients into account along the protein-DNA interface and its based on the interpretation of change in (normalized) binding free energy per unit of change in the shape-parameter value.
A two-step approach has been developed, which takes the quantification of DNA binding specificity based on a strictly mechanism-agonistic model and interpretation in terms of shape readout as a post hoc analysis. With this, a statistical significance to the readout of a particular shape parameter at a particular position within the binding site can be achieved.
![]()
Genomic encoding of transcriptional burst kinetics
In this paper Larsson and colleagues described genome wide characteristics of expressed genes accordingly to two main previously described mechanisms named transcriptional burst frequency and burst size.
But what is a transcriptional burst? In the context of gene expression regulation it refers to how a gene can be expressed. In fact not all the genes are expressed in the same way. Some genes tend to be always expressed, usually housekeeping genes, because they have to assert basic biological functions like regulate the cell cycle for example. Some others, might be important for specific process that happens less frequently like for a key developmental process or during the differentiation of a stem cell population. Hence, these genes will be activated in short time lapses or “pulses” for a multitude of reasons. One of this is defined as transcriptional noise. Transcriptional burst varies in frequency and size but how and which regulatory regions/elements are involved in these two aspects is not completely described.
Larsson and colleagues explored such properties in endogenous human and mouse genes using allele-sensitive single-cell RNA sequencing. What they found was that core promoter elements affect burst size and uncover synergistic effects between TATA and initiator elements. Furthermore, they have found that enhancers control burst frequencies, and demonstrate that cell-type-specific gene expression is primarily shaped by changes in burst frequencies and then mostly mediated by the enhancers.
Larsson, Anton JM, et al. “Genomic encoding of transcriptional burst kinetics.” Nature (2019): 1.
![]()
Using single nucleotide variations in single-cell RNA-seq to identify subpopulations and genotype-phenotype linkage
Single-cell RNA sequencing (scRNA-seq) technology has emerged as a powerful tool to explore cellular functionality at a single cell level and have enabled in-depth interrogation of previously unexplored rare cell types. Pattern of gene expression (GE) is convenient and conventional used as to investigate the cellular function of a cell. When GE combines with single nucleotide variants (SNV), this results in a better identification of cancer cell subpopulations. As SNV identify from scRNA-seq manifest the genetic alterations on gene expression by cis and trans effect.
Poirion et al. (1) introduced a computational framework called Sparse SNV inference to reflect Gene Expression (SSrGE) in order to investigate coupled information from DNA-seq and RNA-seq obtaining filtered, effective and expressed SNVs (eeSNVs). First step in SSrGE is variant calling step that can use GATK pipeline or another preferred SNV calling pipeline. Second, LASSO regression model is used for eeSNVs identification where GE values are considered as a response variable and SNVs as predictor variables (for specific detail please check the publication). The resultant eeSNVs have shown to be more efficient in separating subpopulation than GE alone. eeSNVs identified from scRNA-seq is an efficient way of subpopulation discrimination that can be generalized to non-cancerous cells. The interpretation has a high biological impact because eeSNVs are obtained from scRNA-seq data. The findings from Poirion et al. (1) were supported re-identifying the list of cancer-related genes through SSrGE approach. Moreover, such approach gives a unique opportunity to investigate the relationship between SNV and GE. In summary, one can use SNV features from scRNA-seq data to investigate the genotype-phenotype relationship and subpopulation identification.